 Back Home
Back Home
Some organisms are intrinsically resistant to many (or even all) antimicrobial
agents. Some microbiologists call this "primary resistance". Other organisms acquire resistance, either by mutating or by sharing the
resistance genes of resistant organisms. This should not be confused
with the term "clinical resistance" which refers to the failure of
an antimicrobial to eradicate infection, despite the apparent ability
of the agent to kill the 'bug' in vitro. A wide variety of problems
can account for clinical resistance, such as impaired host immunity,
inadequate drug delivery, and foreign material in the site of a wound.
Antimicrobial resistance is an ever-increasing problem in hospitals, especially
in intensive care units. The problem is so severe that some authorities believe
that we are entering the "post-antibiotic era" where widespread bacterial resistance will render most antibiotics ineffective.
What are the reasons for this resistance?
No-one is sure, but the principle cause appears to be inappropriate and profligate use of antibiotics, especially
in the hospital environment. Several studies have shown widespread misuse of
antibiotics. Another major problem is spread of infecting organisms from patient to patient. This is usually done by the nurses, doctors and others caring for the patient. The major method of spread is on contaminated hands. Over a hundred years after Semmelweiss we still have not learnt the lesson he taught. Semmelweiss reduced mortality rates substantially in his hospital by insisting that students wash their hands before moving from the autopsy room to the maternity wards, and was consequently hounded from office and died (ironically, of septicaemia) in a mental institution.
The following very simplistic picture shows two important features of many bacteria:
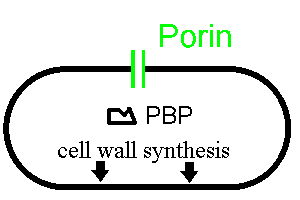
Now let's see how a beta lactam such as penicillin disturbs this process:
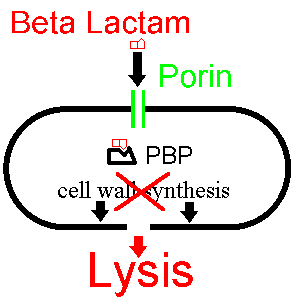
The beta lactam enters the bacterium via the porin and binds to the PBP,
inhibiting cell wall synthesis. The bacterium then dies.
There are several ways that the bacterium can side-step
this attack, and we will use these as a model for all resistance mechanisms, even
in other organisms such as fungi and viruses. Mechanisms are
as follows:
The bacterium on the left has lost the porin that normally transports the antibiotic
into the cell. In a normal environment, such a loss would be a disaster for
the bacterium as it would lose its competitive edge over other bacteria.
Here it is a boon, as it will survive while other porin-rich bacteria will
be killed by the antibiotic!
The bacterium on the right has an even better solution. It has a pump
that pumps out the beta lactam as fast as it comes in. An elegant form
of resistance!
This bacterium has a slightly altered PBP. The PBP can still carry out its
function, but is no longer inhibited by the penicillin, which cannot bind
to it. The bacterium carries on unscathed.
This is an important method of resistance for many bacteria. Beta lactams
are for example broken down by beta lactamases . There is a whole
host of beta lactamases, and new ones seem to be discovered every day.
Although more attention has been focussed on beta lactamases than on
other enzymes that destroy antibiotics, there are many such enzymes that
break down aminoglycosides, chloramphenicol, and so on.
Fortunately for
us, there are several substances such as clavulanic acid that can be
used to inhibit beta lactamases. But the wily bacteria have managed to
get around this too, using a variety of methods!
There are several classifications of beta lactamases. A recent one is that
of Bush, Jacoby & Medeiros { Antimicrob Agents Chemother 1995(39)1211-33 }
which combines features of several other classifications. They define
four groups:
For the fourth method of resistance, we consider another antimicrobial
agent and how bacteria combat it. The agent is trimethoprim .
Bacteria need to make their own folic acid, and they normally do this
using a vital pathway that involves the enzyme dihydrofolate reductase.
Trimethoprim inhibits this bacterial enzyme, preventing folate synthesis
and thus interfering with the ability to make DNA. But some wily bacteria
bypass this step by acquiring a new enzyme that bypasses the old, inhibited
dihydrofolate reductase. The new enzyme comes from (you guessed it) plasmids.
Patients in ICU are often very susceptible to fungal infection, and as
we have gained the ability to sustain the lives of sicker and sicker
people, we have noticed many more fungal infections in ICU. There are
not many antifungal agents, they have been used frequently, and now
we are seeing more and more resistance. The common mechanisms for
resistance in fungi are just those we discussed above: changes in
the binding site of the antifungal agent, and decreased accumulation
of the antifungal within the cell due to decreased permeability and/or
a pump that gets rid of this agent.
Methods for testing in vitro fungal resistance have been argued about
for years. Now National Committee for Clinical Laboratory Standards
macro- and microdilution broth methods have been widely accepted {Alexander
& Perfect, 1997}.
Amphotericin B (AB) is generally an excellent antifungal.
AB binds ergosterol causing leaks in the fungal cell membrane, and may
also stimulate host immunity. Ergosterol-deficient fungi seem to be
resistant to the action of AB. Primary resistance is seen in fungi such
as Pseudallescheria boydii as well as certain species of Candida
e.g. C. lusitaniae. Although some have alleged that prior azole therapy
may deplete fungal membranes of ergosterol and thus decrease the effectiveness
of AB, evidence for this is scanty.
Resistance to azoles such as fluconazole (a "triazole")
can be either due to
acquired fungal resistance to the agent or to acquisition of a different
strain. Long-term fluconazole therapy, especially in AIDS patients is often
associated with resistance. Long-term prophylaxis with this agent may
result in colonisation with intrinsically resistant organisms such as
C. krusei. Azoles act on lanosterol 14-alpha demethylase, inhibiting
ergosterol synthesis. (They may act on other enzymes too). Resistance
to azoles is associated with defects in enzymes on the biosynthetic
pathway for ergosterol, or to decreased intracellular concentrations
of the drugs. Both impermeability and pumps that remove the drug from
the fungal cell have been described. Much work has been done on the
pumps, of which there are two main categories, the ABC and MFS transporters.
(These stand for "ATP-binding cassette" and "major facilitator superfamily").
Azole cross-resistance is probably common.
Fluoropyrimidines such as 5-flucytosine (FC) are useful
antifungals for certain species & strains of Cryptococcus and Candida, and
for chromomycosis. Secondary resistance is common. FC is converted within
fungal cells (but not mammalian cells) to fluorouracil which ultimately
results in abnormal fungal RNA as well as interfering with DNA synthesis.
Loss of one or more of several enzymes may confer resistance to FC.
Newer antifungals such as echinocandin B analogues, and
possibly pradimicins have not yet been thoroughly investigated regarding
fungal resistance.
This is discussed elsewhere
| Date of First Publication: 1999] | Date of Last Update: 2006/10/24 | Web page author: Click here |