and
|
Dr. D. Blythe, FRACP, Senior Registrar Dr. P.V. van Heerden, FFICANZCA, Staff Specialist
Dept. of Intensive Care,
Sir Charles Gairdner Hospital, |
Acknowledgement – This paper is based on a review article published by the authors in Anaesthesia and Intensive Care.
where Ppa = pulmonary artery pressure, Pla = left atrial pressure, and Q = cardiac output. However, "resistance" is a concept derived from the fluid dynamics of laminar, non-pulsatile flow of a Newtonian liquid in channels with a constant radius. Blood is not a Newtonian fluid. Blood flow is pulsatile and turbulent, and the pulmonary vasculature does not have a constant radius. The use of PVR is therefore a simplification and subject to error, but justifiable as the errors become less significant as PVR rises, and the changes seen in pulmonary hypertensive states are of such magnitude that the assumptions made in determining PVR become less important. Nevertheless, estimates of PVR should always be interpreted in the light of supporting clinical information. In order for PHT to develop a number of homeostatic mechanisms in the pulmonary circulation have to be overcome. These mechanisms include -
Table 1. Causes of pulmonary hypertension |
I. Primary pulmonary hypertension II. Secondary pulmonary hypertension
|
Pathophysiological mechanisms that contribute to the development of PHT are hypoxia via the mechanism of HPV, loss of, or mechanical distortion of vessels, or obstruction of vessels by thrombus.
NO has other actions which include –
Inhaled NO has been shown to be an effective SPV in several clinical settings. The first report of its use to decrease PAP was in 1988, when Higgenbottam and co-workers reported that inhalation of NO decreased PAP in patients with PPH.32 The idea was subsequently developed using a number of experimental models.33 PHT was reversed without adverse systemic consequences in the majority of these models, confirming its potential as a SPV. Similar results have been obtained in humans. Rossaint et al.34 reported decreased PAP and improved oxygenation secondary to improved V/Q matching in ten patients with ALI. No adverse consequences were seen. Confirmation of Rossaint’s findings have been reported by Payen,35 and other groups. The focus has now shifted to establishing dose-response relationships and investigating potential toxicity. Of interest is the observation that NO results in improved PAO2 and reduced PA pressures, while cardiac output is unaffected. Intravenous prostacyclin, in contrast, has been shown to reduce PA pressures and increase oxygen delivery, principally by increasing cardiac output. The reasons for these differences in actions are not entirely clear. The initial work on NO has been done with doses ranging from 1 to 128 parts per million (ppm). More recently, using doses of between 60 and 230 parts per billion (ppb) Gerlach et al.36 demonstrated a significant improvement in oxygenation and decrease in shunt fraction without change in pulmonary resistance. Others have also noted that the doses of NO required to improve oxygenation are much lower than the doses required to reduce PAP. This suggests that the dose-response curve for pulmonary artery pressure may not be the same as that for oxygenation, and that the beneficial effect of NO on pulmonary perfusion is due to a redistribution effect, rather than pure enhancement of flow. In most cases, in adults, concentrations of 5-40ppm are sufficient to lower PAP. Concentrations as low as those used by Gerlach approach the level found in room air (usually around 10 ppb although levels in expired air can reach 250ppb). The response to NO is not entirely predictable. Several studies have now documented groups of critically ill patients with ALI who have not responded to low-dose NO (<40ppm).34,37 The mechanism of this variability in response to NO is not known, although the degree of initial PHT appears to correlate with the likelihood of response, those patients with the most marked PHT being most likely to benefit.37 Withdrawal of NO therapy has also proven to be problematic in patients with ALI and PHT. Sudden worsening of PHT and consequent hypoxaemia has been reported on cessation of therapy.37 This is an unpredictable phenomenon and the mechanism is not known. Possibilities include suppression of endogenous NO production by exogenous administration (product inhibition), or release of a vasoconstrictor substance. The toxicity of NO remains an area of active research. NO reacts with oxygen to form strong oxidants with as yet incompletely understood effects on normal and pathological cellular metabolism. The rate of oxidation is dependent on the oxygen concentration and the square of the NO concentration.91 At high concentrations, NO causes systemic methaemoglobinaemia and has caused death.38 At lower concentrations, the avid binding to haemoglobin may protect the systemic circulation from toxicity, but the pulmonary circulation and alveoli remain at risk. The clinical significance of reduced platelet adhesion following NO administration is not clear. 39 There are no reports of coagulation problems with NO in clinical use. Most toxicological studies have not addressed the issue of long-term NO administration, or the potential for accumulation over prolonged periods of therapy. There are studies reporting NO administration to infants for periods of up to several weeks, 40 and to adults with PPH in a pulsed fashion over periods of up to nine months, without adverse effect.41 However, the higher oxides of nitrogen are known to be toxic substances. Nitrogen dioxide (NO2) has considerable pulmonary toxicity, causing pneumonitis and pulmonary oedema, and has been reported as a cause of cytotoxicity and death in experimental animals.42.43 NO2 is metabolised to nitrous and nitric acids in aqueous solution, and a number of other higher nitrates and nitrites are formed with as yet unknown effects.44 Metabolism of NO to NO2 is therefore a potential hazard, and NO and NO2 monitoring are mandatory during use of NO, so that ambient levels of NO do not exceed 25ppm, nor NO2 levels exceed 1ppm. 45 Apart from potential toxicity, other problems with administration of NO include the delivery system required for its use and the high capital costs of delivering the agent. Although NO gas itself is currently not expensive, the cost of administration is significant.
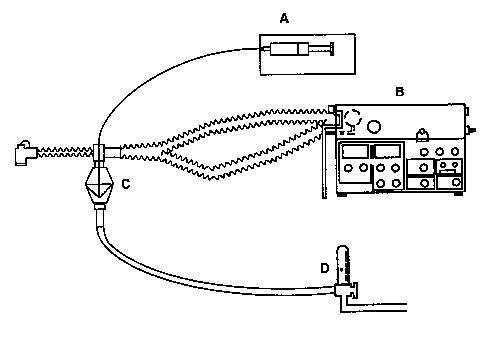
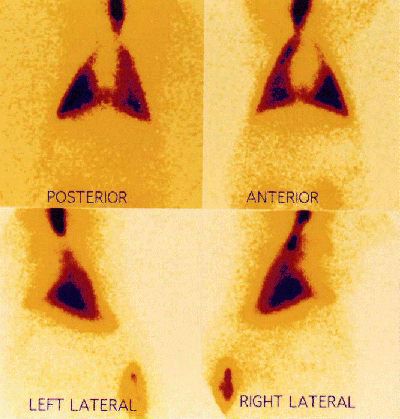
The actual amount of drug deposited in the alveolus by such apparatus remains uncertain. A distal site of deposition is inferred from a knowledge of the particle size delivered and from the clinical effect observed. To achieve distal deposition a particle must be of size 0-5 µm.57 Even with appropriately-sized particles, the amount of a nebulised drug which actually reaches the alveolus in a ventilated patient is known to be a small fraction (less than 10%) of the total amount administered.65-7 The authors have performed nuclear medicine ventilation scans in piglets, using radiolabelled DTPA dissolved in the prostacyclin glycine buffer to determine site of drug deposition.68 A typical ventilation scan is shown in figure 2, indicating widespread and distal deposition of the tracer when delivered by jet nebuliser.
As regards toxicity of IAP, systemic haemodynamic side-effects (hypotension, tachycardia) have not been seen with IAP. PGI2 is not transformed into an active form in the lung, nor is it significantly metabolised.69 Arterial concentrations of its major metabolite,
6-keto prostaglandin F1
![]() have usually been reported as unchanged during inhalation of PGI2.55,70 Recent unpublished work from our institution describes arterial levels of
6-keto prostaglandin F1
have usually been reported as unchanged during inhalation of PGI2.55,70 Recent unpublished work from our institution describes arterial levels of
6-keto prostaglandin F1
![]() in excess of 500 pg/ml during IAP therapy. These levels of
6-keto prostaglandin F1
in excess of 500 pg/ml during IAP therapy. These levels of
6-keto prostaglandin F1
![]() have been shown to have a significant antiplatelet effect in vitro. The major area of concern has been the effect of IAP on coagulation.71 There has been no report of any clinically-important adverse effect on coagulation using PGI2 via the inhaled route. No other systemic toxic effects have been described. PGI2 has been reported as being irritant to the airway when administered intratracheally.72 A final area of concern is the alkalinity (pH 10.5) of the glycine buffer medium in which PGI2 is prepared. The authors have reported acute inflammation of the airways of piglets to which high dose IAP had been administered. 68
have been shown to have a significant antiplatelet effect in vitro. The major area of concern has been the effect of IAP on coagulation.71 There has been no report of any clinically-important adverse effect on coagulation using PGI2 via the inhaled route. No other systemic toxic effects have been described. PGI2 has been reported as being irritant to the airway when administered intratracheally.72 A final area of concern is the alkalinity (pH 10.5) of the glycine buffer medium in which PGI2 is prepared. The authors have reported acute inflammation of the airways of piglets to which high dose IAP had been administered. 68
| Date of First Publication: 1999/2/22 | Date of Last Update: 2006/10/24 | Web page author: Click here |