What is autoregulation?
If the pressure perfusing the arteries of almost any organ is varied, flow through the organ changes very little. This is termed autoregulation. Autoregulation only occurs between certain pressure limits - if the pressure drops too low or soars too high, autoregulation fails, and organ perfusion is compromised - at low pressures, perfusion drops, and at high pressures, excessive flow occurs. It used to be thought that flow was constant throughout the 'autoregulatory range', and then dropped off at a precise lower threshold, the 'lower limit of autoregulation' often abbreviated to LLA. More recent and general work (the initial clinical report was based on one woman with pregnancy-induced hypertension!) suggests that this is simplistic, and that the curve, well, curves as it nears the LLA (and indeed upper limit).
Practically, this means that we should define the LLA as the point where, as the perfusion pressure drops, the flow drops by a pre-defined value. In other words, autoregulation is starting to fail at a pressure where flow drops by say ten percent.Gao and colleagues have recently reviewed autoregulation in perhaps the most important organ of the lot - the brain. Cerebral autoregulation has been extensively studied, but unfortunately all the studies don't agree. Gao describes three models of autoregulation:
- 'Type 1' - flow depends only on pressure below the LLA and above the upper limit (ULA). Implicit in this model is that the vessels are maximally vasodilated below the LLA, and maximally vasoconstricted above the ULA. This in turn implies that the slope of the flow:pressure curve above the ULA is less steep than the slope below the LLA.
- 'Type 2' - similar to Type 1, but above the ULA the slope of the flow:pressure line is the same as the slope below the LLA. The implication of this model (designed to account for the experimental observations) is that above the ULA flow increases more than would be expected if the flow:pressure relationship was simply passive.
- 'Type 3' - A complex mathematical relationship between flow and pressure. One possible curve is:
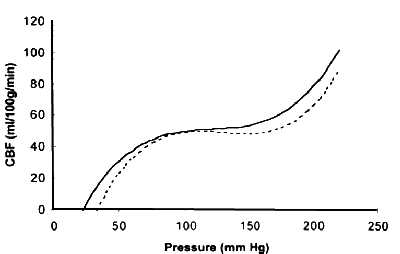
This last option is almost certainly the 'correct' one. Dirnagl and Pulsinelli have fitted experimental curves (from rats) using a third-order polynomial, but even this is probably simplistic. Gao derives a more complex, four-compartment model, and wisely points out that even this model has serious limitations.
In summary, autoregulation is complex, and difficult to model. Human studies of cerebral autoregulation have (for obvious reasons) generally avoided exceeding the upper limit. Such studies have generally found the LLA in brain to be 70 to 93 mmHg. Average values from all the studies reviewed by Gao were 80 mmHg for the LLA. The single study that allowed a guess at the ULA put this at 161 mmHg, but hypertensive persons were included. This is important, as there is evidence that in people with longstanding hypertension, both ULA and LLA are reset to higher values.
Does autoregulation vary from organ to organ?
Mechanisms of autoregulation may vary substantially between organs. There is evidence that coronary autoregulation is, for example, quite different from brain autoregulation.
During haemorrhagic shock (in pigs) microcirculatory blood flow in the stomach, colon, liver and kidney decreased in concert with lowering of systemic blood flow; flow in the jejunal mucosa was preserved, and pancreatic blood flow was selectively impaired! [Br J Anaesth 2000 Apr;84(4):468-75]
Experimentally, differential autoregulation has also been shown in response to increases in blood pressure. For example, in response to infusion of pressor agents, renal autoregulation is almost unimpaired, while regulation in the mesenteric vascular bed is less good, and differs with the different agents.
In the following sections, we briefly look at the various circulations.
Brain autoregulation
We have already mentioned the rather inadequate models that have been constructed of brain autoregulation. We will not delve deeply into these, as they are generally empiric, with little grounding in physiology.
Of much more interest is recent work on the underlying physiology of autoregulation in the brain. The main regulator of brain blood flow is pressure-dependent activation of smooth muscle in the arterioles of the brain. The more the arteriole is stretched, the more it contracts, and this lasts as long as the stretch occurs.
Let us consider in more detail what happens when arteriolar muscle is stretched. The sequence of events is as follows:
- Increased perfusion pressure causes increased stretch ; then
- Phospholipase C is activated, resulting in diacylglycerol production, which causes arachidonic acid release;
- Cytochrome P450 4A2 (CYP 4A2) converts the arachidonic acid to..
- 20-hydroxy eicosatetraenoic acid (20 HETE ), which in turn..
- activates protein kinase C; this activation
- Inhibits potassium channels, especially the calcium-activated potassium channel called K Ca , resulting in..
- A lowering of the membrane potential of the vascular smooth muscle, with
- Increased smooth muscle activation (probably mediated through a rise in intracellular calcium levels), causing vasoconstriction .
The above complex sequence of events is only half the story! It is clear that, although the above pressure-dependent activation will maintain flow over a wide range of pressures, what if local nerve cell metabolic activity increases? How does the brain 'shunt' extra blood to an area that needs it?
An intriguing answer is contained in the following scenario:
- Glutamic acid (glutamate ) spills over from metabolically active neurones. This
- induces astrocyte membranes to release arachidonic acid;
- The arachidonic acid is converted by Cytochrome P450 2C11 (in rats) to epoxyeicosatrienoic acid (EET );
- EET diffuses to nearby smooth muscle and stimulates K Ca , antagonising the effect of HETE.
Cerebral autoregulation can thus be seen as a dynamic balance between HETE, which tends to constrict vessels, and EET, which dilates them! Note that with (for example) hypoxia, other metabolites would also tend to increase cerebral vasodilatation. Even the above picture is simplistic, as astrocytes may release other potentially vasoactive signals, such as thromboxane A2, PgE and PgF, and prostacyclin. K Ca channels may be affected by a host of mediators, including nitric oxide, extra-cellular K +, adenosine, and prostacyclin. Regulation of cerebral autoregulation is summarised in the following diagram (after Harder et al, 1998).
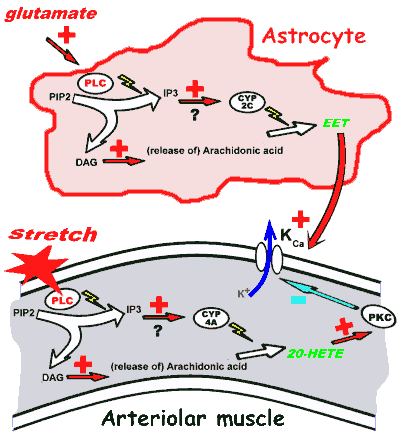
| Cerebral autoregulation. Muscle stretch activates phospholipase C (PLC), resulting in release of arachidonic acid and its conversion into 20-HETE, which inhibits K Ca , resulting in membrane depolarisation, increased intracellular calcium concentrations (not shown), and muscle contraction. This is antagonised by EET, which is produced in an analogous fashion when glutamate that spills over from metabolically active neurones activates PLC in astrocytes. |
Cerebral autoregulation may be quite heterogeneous. For example, autoregulation in small brainstem vessels may be dependent on K ATP !
Renal autoregulation
In some ways this resembles autoregulation in the brain. The main site of autoregulation in the kidney is however the afferent glomerular arteriole. There are two main factors that affect vascular tone in the afferent arteriole:
- Stretch-activated constriction of vessels (as for brain);
- 'Tubulo-glomerular feedback'.
Autoregulation in the kidney is said to occur between about 80 mmHg and 180 mmHg (Navar, 1997). Both of the above mechanisms are important to maintenance of near-constant blood flow. Stretch-activated vasoconstriction appears very similar to that in the brain - stretch results in membrane depolarisation, increased intra-cellular concentrations of calcium ions, and ultimately, vasoconstriction. Again, phospholipase C activation is important.
Tubulo-glomerular feedback (TGF ) is even less well understood. Complex signals pass from the macula densa to the afferent arteriole, regulating its tone. The fundamental theme of TGF is that increased delivery of fluid and/or NaCl to the distal tubule causes vasoconstriction , thus limiting the flow (negative feedback). Signals between macula densa and arteriole may include:
- Angiotensin II, which modulates TGF by stimulating AT 1 receptors but is not the primary mediator of TGF (its effects oppose TGF);
- nitric oxide (made by the neuronal form of nitric oxide synthase, present in the macula densa) - this seems to dampen the TGF mechanism, not to mediate it!
- Prostaglandins and thromboxane also modulate TGF (Cyclo-oxygenase inhibitors alter the autoregulatory response in the kidney);
- Cytochrome P450 metabolites, notably 20-HETE, are probably important in vasoconstrictor components of TGF;
- Stimulation of A 1 adenosine receptors by adenosine released from the macula densa may mediate afferent arteriolar vasoconstriction;
- ATP , perhaps the most important mediator of TGF, specifically by stimulating a ligand-gated ion channel that depolarises smooth muscle cells of the afferent arteriole (P 2 receptors)!
Renal autoregulation is complex and challenging, with numerous complementary mechanisms maintaining renal blood flow and glomerular filtration rate over a wide range of pressures.
Coronary autoregulation
Control of coronary blood flow differs depending on the type of vessel being considered - arteries, large arterioles or smaller arterioles. Coronary capillaries also appear to make a significant contribution to coronary vascular resistance! As if this is not enough, transmural variations occur.
Coronary vessels have intrinsic (myogenic) control systems. Potassium
channels are important here, but in contrast to the cerebral vasculature,
these appear to be K ATP channels
(blocked by glibenclamide).
The major factor controlling
the resistance of the coronary microcirculation appears to be local
production of vasodilatory metabolites.
Neither K ATP nor adenosine (see below) seem vital to
the preservation of coronary autoregulation, at least between
perfusion pressures of 60 and 100 mmHg.
Interestingly, myocardial oxygen consumption seems to vary with coronary perfusion pressure and, more importantly, with coronary flow (Gregg's phenomenon). In other words, greater coronary blood flow increases myocardial metabolism! This phenomenon antagonises autoregulatory controls.
Local myocardial adenosine production appears important as a regulator of coronary flow. Ischaemic myocardium releases increased amounts of adenosine and inosine, for example. The coronary endothelium also appears to play an enormous part in regulating coronary arterial tone. Endotoxin administration (in rats, at least) appears to cause massive coronary vasodilatation, probably mediated by nitric oxide. Endothelial NO production in the coronary circulation seems to mediate coronary vasodilatation in response to adenosine infusion. Coronary endothelial cells also appear to have the capacity to produce epoxyeicosastrienoic acids, which cause coronary vasodilatation.
'Circulatory reserve' in the heart is very important because of the near-maximal extraction of oxygen from coronary blood at rest. Normal hearts appear to have a four or five-fold reserve (ability to increase flow).
Muscle autoregulation
An important feature of resting skeletal muscle is that it appears to be substantially vasoconstricted. This has been attributed to high baseline sympathetic tone - 20 HETE may also play a role. During exercise, accumulation of local metabolites appears to mediate increased blood flow and thus, oxygen delivery. Such reactive hyperaemia may be impaired in disease states, for example in cardiac failure.
Splanchnic autoregulation
It has been said that total intestinal blood flow is autoregulated between pressures of about 80 mmHg and 160 mmHg, with autoregulatory adjustments occurring in small pre-capillary vessels. Villous vascular autoregulation is said to be better than in vessels supplying the intestinal musculature, but with dropping pressures the efficiency of the intestinal countercurrent exchanger increases, putting villi at risk for hypoxia. [J Hypertens Suppl 1989 Sep;7(4):S79-84]
Part of the normal regulation of mesenteric vascular tone in the rat (and probably man) appears to be modulated by alpha-1 adrenergic mechanisms. There is also good evidence for a mesenteric arterial baroreflex (in rats, anyway). Sympathetic outflow (via noradrenaline release) may regulate the basal vascular tone, but many other factors may modulate this effect, including non-adrenergic, non-cholinergic nerves and the parasympathetic nervous system. In addition to alpha-1 adrenergic effects, some evidence suggests that postjunctional alpha-2 receptors cause vasoconstriction.
An intestinal vascular myogenic response appears to be present. An interesting study by Mackay and colleagues [Crit Care Med 1995 Jun;23(6):1090-8] in pigs on normothermic cardiopulmonary bypass showed that autoregulatory thresholds were higher in the kidney and splanchnic organs than in the brain. Pump pressures of 45 mmHg were associated with splanchnic and renal hypoperfusion, but the brain appeared to autoregulate adequately. Of great importance is the observation that (in contrast to increasing pump perfusion pressures), administration of dopamine had no beneficial effect on perfusion of the poorly-perfused organs. Despite differences in the magnitude of the response from that in cerebral vessels (rat, rabbit), changes in intracellular calcium concentrations in response to pressure changes appear similar to those seen in the cerebral vasculature.
The bowel vasoconstrictor response to angiotensin II (but not that seen with noradrenaline) appears to be mediated by 20-HETE. This vasoconstriction is blocked by the selective CYP450 4A inhibitor, DDMS (N-Methylsulphonyl-12, 12-dibromododec-11-enamide).
Note that in the rat, after initial vasoconstriction in response to noradrenaline infusion, the bowel partially escapes ! Multiple factors are probably important in this escape, which is almost totally abolished by the combination of adenosine deaminase and propranolol. Nitric oxide may play a role in mediating the escape phenomenon. The antagonism of SMA autoregulation by nitric oxide is well-documented. It has also been suggested that increasing local hydrogen ion concentrations contribute to the 'escape' by decreasing the sensitivity of post-junctional alpha-2 receptors to noradrenaline.
References
- Harder DR et al. Acta Physiol Scand 1998 164 527-32. A complex review of the role of P450 in cerebral autoregulation.
- Gao E et al. Am J Physiol 1998 274 H1023-1031. A somewhat empirical model of cerebral autoregulation, with a good review of previous studies.
- Navar LG Am J Physiol 1998 274 F433-444. A good summary of the complexities of renal autoregulation.
- Gebremedhin D et al, Circ Res 2000 87 60-5. An investigation (in rats) into the role of HETE in autoregulation. CYP 4A1,2,3 and 8 appear to be involved.
- For the enthusiast, numerous references are embedded throughout the document as a sub-text (Click on View | Source in your browser).
| Date of First Publication: 2000-7-16 | Date of Last Update: 2006/10/24 | Web page author: Click here |