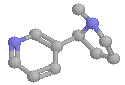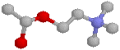
From our previous tutorial on the adrenergic component of the ANS, you will now be thoroughly familiar with CHIME (heh)! Here's the acetylcholine molecule:
Synthesis of acetylcholine (ACh) is simple:
acetyl-Coenzyme A + choline ==> acetylcholine
The enzyme catalysing this reaction is choline acetyltransferase , found both in mitochondria and the cytosol. There are some similarities between noradrenergic synapses (discussed in the previous web-page) and cholinergic ones. Remember that at noradrenergic synapses, about 50% of noradrenaline is taken up by a presynaptic transporter? At cholinergic synapses there is also a presynaptic transporter - the difference is that ACh is first hydrolysed back to acetate and choline, and then choline is avidly taken up. It is this presynaptic uptake of choline that is the main regulator of ACh synthesis.
Release of ACh follows the pattern we have come to know - ACh is concentrated in large amounts in presynaptic vesicles, which release their contents into the synapse when voltage-gated calcium channels open in response to membrane depolarisation. The release and effects of ACh have been characterised in minute detail, especially at the neuromuscular junction where far larger amounts of ACh are released than at other sites. (We will not discuss neuromuscular transmission in detail, as we are concentrating on the autonomic nervous system, although exam candidates would be well-advised to acquaint themselves with the minutiae)!
As we would expect, there are specific transporter proteins involved in moving both ACh and choline around:
One cannot over-emphasise the importance of cholinesterase , the enzyme that breaks down ACh at the synaptic cleft. Cholinesterase inhibitors are important both as toxins, and as therapeutic agents.
We will here concentrate on acetylcholine actions in the peripheral nervous system. Acetylcholine acts on two vastly different classes of receptors - nicotinic receptors (with two subtypes, one at the neuromuscular junction of skeletal muscle, the other within ganglia and the CNS), and muscarinic receptors (widely distributed within both peripheral and central nervous systems). Some drugs other than ACh also stimulate both nicotinic and muscarinic receptors, for example, carbamylcholine and carbachol.
Note that the effects of cholinergic agents are complex, not only because of the existence of nicotinic and muscarinic receptors (and subclasses of these) but also because of the presynaptic and postsynaptic locations of such receptors. To make things even more complex, ACh has inhibitory (heterotropic) effects on sympathetic neurones!
As an aside, the bite of the black widow spider (Latrodectus) is so toxic because of massive release of acetylcholine from neurones. This is mediated by alpha latrotoxin, which appears to conduct calcium ions into the presynaptic terminal.

The prototype drug that stimulates nicotinic receptors is of course nicotine, which stimulates the nicotinic receptors on muscle cells, those found within autonomic ganglia, and (not least) nicotinic receptors within the nervous system.
Some drugs that stimulate nicotinic receptors also cause block, for example nicotine itself. The mechanism of this block is instructive - ongoing stimulation of the receptors causes decreased electrical excitability due to inactivation of voltage-sensitive sodium channels.
Another, even more interesting block occurs. This "Phase II block" is seen clinically in skeletal muscle with repeated administration of doses of suxamethonium - the receptors become desensitised to stimulation, one of the several reasons not to give a second dose of 'sux'. Phase II block also occurs with nicotine stimulation of receptors.
We will not dwell on the nicotinic receptors on muscle cells, concentrating instead on the neuronal type, found in both sympathetic and parasympathetic ganglia. Nicotinic receptors are ligand-gated ion channels - unlike the fairly stereotyped muscle receptor, the neuronal one is quite heterogeneous, being made up of a mixed bag of a variety of different alpha and beta subunits. (There seems to be little clinical significance of this mix, at present). ACh binds the alpha subunit and this results in opening of an ion channel.
Agonists such as nicotine are mainly toxicological curiosities, rather than clinically useful
drugs. Epibatidine (from the Dendrobatid poison dart frog Epipedobates tricolor ) is a potent
agonist at nicotinic receptors. Interestingly enough, in the CNS, profound analgesia is induced,
but peripheral effects of course preclude its use as an analgesic - it binds not only
nerve but also muscle nicotinic receptors. More nerve-specific analogues have been
synthesised.
Phase II trials of the epibatidine analogue ABT-594 are now being started.
Here's
 more than you ever wanted to know
about epibatidine, and here's the molecule:
more than you ever wanted to know
about epibatidine, and here's the molecule:

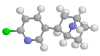
Other nicotinic agonists are lobeline (from the Lobelia plant), and dimethylphenylpiperazinium (DMPP), both of which favour ganglionic receptors, although the former is pretty nonspecific. Nicotine itself slightly favours nerve receptors over muscle ones.

Ganglionic transmission of impulses is insanely complex. For example, in sympathetic ganglia, physiologists have observed four distinct responses to stimulation, with different onset and duration of effect. There are several ways in which drugs can block ganglionic transmission. Here are a few:
The first ganglionic blocker was hexamethonium - it's now merely a curiosity, made
more interesting by Paton's quaint description of the "hexamethonium man" - usually
enthusiastically reproduced in pharmacology textbooks. We won't repeat it
here.
Interestingly enough, hexamethonium and related agents don't interfere with
the ACh receptor directly, in fact, they block the ion channel.

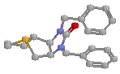
Trimetaphan appears to work by competitive block of ACh receptors. Mecamylamine is also a blocker of neuronal nicotinic receptors, initially developed as an antihypertensive ganglionic blocker, abandoned because of postural hypotension, and now of interest because its noncompetitive voltage-dependent interference with nicotinic receptors makes it significant in the study of addiction. (And you wondered why cigarettes are addictive)! In contrast, agents such as alpha-bungarotoxin, obtained from the venom of an Indian snake called the krait, specifically block muscular nicotinic receptors.

Alpha bungarotoxin was pretty important in the initial isolation and characterisation of muscular ACh receptors. In contrast, the original non-depolarising neuromuscular blocking agents that were used clinically were far from specific for muscular nicotinic receptors. Tubocurarine, for example, also causes significant ganglionic blockade. (As do the similarly obsolete drugs gallamine and decamethonium).

These receptors are peculiar - one might expect them to be responsible
for inhibitory feedback at nicotinic synapses, but they appear to
do just the opposite! This is particularly impressive at the neuromuscular
junction, where they increase acetylcholine release with repeated
stimulation - and are thus important in tetanic stimulation.
The central effects of nicotine may be mediated by its stimulatory effect
on presynaptic nicotinic receptors.
ACh has similar agonist effects at both muscarinic and nicotinic receptors,
but there are several other agents that favour muscarinic receptors.
Muscarinic receptors all mediate their effects through G proteins.
From our knowledge of muscarinic effects on various organs, we can
work out the clinical manifestations of such agents — miosis, sweating,
salivation, abdominal cramps, and often, bradycardia.
The prototype agonist is muscarine, derived from the poisonous fly agaric,
Amanita muscaria.
There are many other muscarinic agonists, including pilocarpine,
bethanechol, and methacholine, but they have few medical uses.
Methacholine (which is simply ACh with a methy group on the choline)
has been used in carefully titrated amounts to diagnose
asthma. Pilocarpine is still used in glaucoma (and has been
used for treatment of Sjogren's syndrome);
bethanechol, the carbamate derivative of methacholine,
has been used in the past to goad the lethargic bladder
or gastrointestinal tract into activity.
Pharmacologists like to rhapsodise
about "structure-activity relationships" and these agents haven't escaped
their scrutiny. The activity of ACh depends on a positively-charged
quaternary ammonium group, and an atom capable of forming a hydrogen
bond through an unshared pair of electrons. The distance between
the two may determine whether agents are nicotinic or
muscarinic.
Tweaking the ester lowers hydrolysis by cholinesterase (most of the above agents
are not/minimally hydrolysed by cholinesterase).
Pilocarpine, unlike many of the other (quaternary) agents which penetrate
membranes poorly, has a tertiary amine group,
so it can penetrate the eye when applied topically. Muscarinic agonists
within the eye have several effects, causing miosis and ciliary
muscle contraction, and indirectly improving drainage of aqueous
humour through the canals of Schlemm.Muscarinic receptors and drugs


Although five muscarinic receptors have been identified, helpfully labelled M1 to M5, only three are well-characterised. (M4 receptors are confined to the CNS, and M5 are not well understood). Here are the three important ones:
M5 receptors seem similar to M1 and M3 in their effects; M4 are similar to M2.
Muscarinic antagonists are medically important. The traditional antagonists (atropine and hyoscine) are not specific for particular receptor subtypes. There are selective agents - the infrequently used anti-ulcer drug pirenzepine is M1 selective (you can work this out once you know it inhibits acid secretion), and gallamine, quite apart from its other effects, is M2 selective. The unpronounceable hexahydrosiladifenol is an M3 specific antagonist. (There are others: dicyclomine is M1 specific, "AF-DX 116" blocks M2).
Detailed knowledge of atropine , scopolamine (=hyoscine) and the synthetic drug glycopyrrolate is vital for all anaesthetists (and examination candidates)! The major differences between atropine and glycopyrrolate are:
One peculiar response to atropine is the bradycardia reportedly seen with small doses (eg. 0.4 mg in an adult) - the mechanism of this paradoxical effect is still not clear, if it exists at all!

Scopolamine has pronounced CNS sedative effects, and may also be useful in preventing nausea and vomiting, especially due to motion sickness.

Ipratropium is not without interest, as it appears to be a fairly useful agent in the management of bronchospasm, especially in those with chronic obstructive airways disease with some airway hyperreactivity. Because ipratropium is quaternary, it is very poorly absorbed when delivered as an aerosol, confining its effects to the airways. In addition, ipratropium does not inhibit mucociliary clearance (an unfortunate side-effect of atropine therapy).
The effects of poisoning with antimuscarinics are largely predictable from a knowledge of muscarinic receptors and their location - dry mouth, impaired sweating, mydriasis, and tachycardia, as well as profound CNS dysfunction with delirium and hallucinations. These effects are far more dangerous in children than in adults. The mnemonic "red as a beet, blind as a bat, dry as a bone, mad as a hatter, and hot as hell" is apt.
Other agents with anticholinergic activity include tricyclic antidepressants, some major tranquillizers, and many of the older ('sedating') anti-histamines.
We have already listed some non-selective muscarinic agonists. There are a few muscarinic agonists that are selective for certain sub-types of muscarinic receptor, but they are of little clinical significance. Oxotremorine and "McNA343" are M1-selective.
The main drugs of clinical importance that interfere with ACh metabolism are the anticholinesterases. There are several other drugs that interfere at other points in ACh metabolism, but apart from botulinum toxin, they have little clinical relevance. For the record, here are some:
 Acetylcholinesterase (ACHE) has two sites important in hydrolysis
of ACh - the anionic site and esteratic site. There are several
classes of agent that inhibit ACHE, raising ACh levels in the synapse.
Cholinesterase inhibitors bind the esteratic site, usually to an
acetate, carbamate or phosphate group. Clinically the most used is undoubtedly
neostigmine, given as reversal of neuromuscular blocking agents
administered during anaesthesia. The anticholinesterases may be:
Acetylcholinesterase (ACHE) has two sites important in hydrolysis
of ACh - the anionic site and esteratic site. There are several
classes of agent that inhibit ACHE, raising ACh levels in the synapse.
Cholinesterase inhibitors bind the esteratic site, usually to an
acetate, carbamate or phosphate group. Clinically the most used is undoubtedly
neostigmine, given as reversal of neuromuscular blocking agents
administered during anaesthesia. The anticholinesterases may be:
All of the organophosphates irreversibly bind ACHE. Pralidoxime has been widely used early on in organophosphate poisoning to 're-constitute' the ACHE molecule, although its effectiveness has recently been called into question. Obidoxime has similar effects:
![]()
Physostigmine is a short-acting cholinesterase inhibitor that has been used in the past to manage atropine toxicity (especially in children, where atropine toxicity may be life-threatening). It has good CNS penetration, but a short half-life, and nicotinic effects should be carefully monitored.

comprehensive review
of neuronal nicotinic receptors.
Thanks to Jet for picking up an error under "Muscarinic Receptors".
| Date of First Publication: 2002/7/16 | Date of Last Update: 2006/10/24 | Web page author: Click here |